Primary Analysis
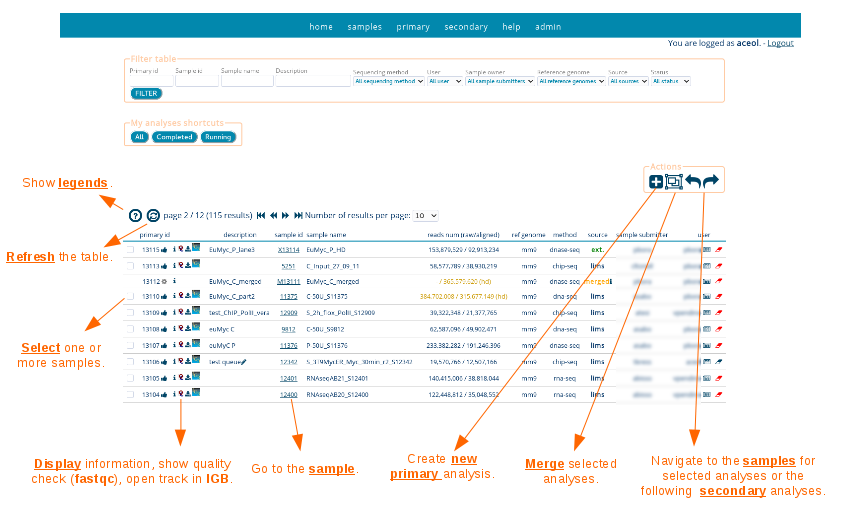
Primary analyses can be submitted on a sample or on a group of
samples. A primary analysis consists of filtering, quality control
and alignment to reference genome of the reads from a specific
sample.
The sequencing technologies currently accepted by HTS-flow are
- RNA-Seq,
- ChIP-Seq,
- DNaseI-Seq,
- BS-Seq.
Several options are applicable to primary analyses:
- Remove Bad Reads: removal of reads that has been labelled bad from the sequencer (grep -A 3 '^@.* [^:]*:N:[^:]*:' | grep -v -- '^--$' | sed 's/ [0-9]:N:[0-9]*:[A-Z]*$//g').
- Trimming: trim the reads starting at 5' if nucleotide quality Q is below 20. Phred quality scores Q are defined as a property which is logarithmically related to the base-calling error probabilities P. To be used if exists the possibility of high degradation of quality at 5' ends of reads.
- Masking: mask nucleotides along the whole reads with N if their Q quality score is below 20.
- Program: tophat/bwa/bismarck - tophat is used for aligning RNA-Seq reads, bwa for ChIP-Seq and DNaseI-Seq, bismarck for BS-Seq.
- Alignment Options: this line is intended for changing the options provided to the aligners.
- Paired: for both tophat and bwa the set of options that treats paired-end reads.
- Remove Duplicates: the final alignment file is processed for removing PCR duplicates ( reads that align on the same genomic location ).
Merging primary analysis
It is possible to select a group of aligned samples and pool their
reads to obtain a merged alignment file. To be merged, the samples
need to be aligned to the same reference genome. This function is
available from the primary analysis page, with the following
parameters.